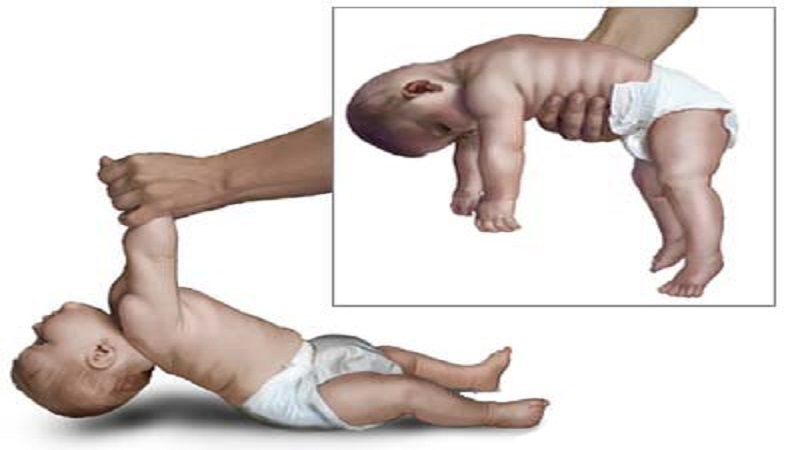
بیماری آتروفی های عضلانی نخاعی
بیماری آتروفی های عضلانی نخاعی، ژنتیکی می باشد و به صورت صفت اتوزوم مغلوب مندلی به ارث می رسد و قابل تشخیص در آزمایشگاه ژنتیک می باشد. آسیب پاتولوژیک رخ داده در SMA در نتیجه تداوم روند مرگ سلولی برنامه ریزی شده (آپوپتوز) است که در دوران جنینی به طور طبیعی وجود داشته است. نوروبلاستهای حرکتی اضافی و دیگر نورونها از نورواکتودرم اولیه تولید می شوند ولی فقط نیمی از آنها زنده مانده و بالغ شده و تبدیل به نورون می شوند. بقیه سلول ها چرخه زندگی محدودی داشته و دژنره می شوند. اگر روندی که به طور فیزیولوژیک باعث توقف مرگ سلولی میشود مختل شود و نتواند در مرحله مشخصی عمل نماید، مرگ نورون ها ممکن است تا اواخر دوران جنینی و حتی بعد از تولد ادامه ی ابد.

انواع بیماری آتروفی های عضلانی نخاعی
یک فرم شدید شیرخوارگی به نام بیماری وردنیگ – هافمن يا SMA نوع 1، یک فرم دیررس شیرخوارگی با پیشرفت آهسته تر به نام SMA نوع ۲، و یک فرم مزمن تر یا جونایل به نام بیماری کوگلبرگ – ولاندر یا SAMIA نوع ۳. یک فرم شدید جنینی که معمولا در دوره پریناتال کشنده است، به نام SMIA نوعه 0 صفرشناخته شده که دژنراسیون نورون های حرکتی در نخاع از اواسط دوران بارداری شروع می شود.
این تقسیم بندی بالینی بوده و براساس سن شروع، شدت ضعف و سير بالینی انجام می شود. بیوپسی عضله انواع ۱ و ۲ را از هم افتراق نمی دهد ولی نوع ۳، الگوی بالغ تری نسبت به الگوی پریناتال دنرواسیون وعصب دهی مجدد را نشان می دهد. در نوع ۲، نمای بیوپسی، شبیه میوپاتی میوتوبولار است به علت وجود توقف تکاملی. میوتیوب های پراکنده و سایر فیبرهای جنینی نارس که در بیوپسی عضله بیماران با نوع ۱ و ۲ هم دیده می شوند ولی غالب نیستند.
حدود ۲۵% بیماران مبتلا به نوع ۱ هستند.
۵۰% به نوع ۲ و ۲۵٪ به نوع ۳ بیماری مبتلا هستند. نوع نادر بوده و کمتر از ۱٪ موارد را شامل می شود. بعضی بیماران از نظر عملکرد بالینی بین نوع ۱ و ۲ یا بین نوع ۲ و ۳ قرار دارند. یک واریانت SMA، به نام بیماری فازيو – لونده، فلج بولبار پیشرونده ای است که ناشی از دژنراسیون نورونهای حرکتی، بیشتر در ساقه مغز می باشد تا نخاع ولی اعصاب کرانیال که به عضلات خارج چشمی عصب دهی می کنند در این نوع هم درگیر نمی شوند.
نورونهای حرکتی اتونومیک سمپاتیک و پاراسمپاتیک نیز درگیر می شوند، اما معمولا تا مراحل پایانی بیماری، تظاهرات بالینی نشان نمی دهند. نقایص اتونومیک ممکن است در تمام انواع SMA عضله دترسور مثانه یا عضله صاف اسفنکترهای اورترا و مقعد را درگیر نمایند. در برخی از بیماران مبتلا به SMA نوع او دیسترس تنفسی ممکن است اختلال شدید در تنظیم اتونوم همراه با دیس اتونومی و کلاپس قلبی عروقی دیده شود که منجر به مرگ و یا آسیب مغزی ایسکمیک شدید شود.
ژنتیک بیماری
هر سه تیپ این بیماریوراثت مغلوب اتوزومی را نشان می دهند. شکل های متعدد بسیار نادرتر آن با بروز دوران بلوغ و دیرتر نیز توصیف شده است که ممکن است الگوی وراثت غالب اتوزومی داشته باشند.
تیپ ۱ معمولاً درجه بالایی از همسازی درون خانوادگی نشان داده، خواهر برادرهای مبتلا یک خط سیر بالینی تقریباً یکسانی را به نمایش می گذارند. در تیپ ۲ و۳، گوناگونی درون خویشاوندی می تواند کاملا مشخص باشد.
در همه ی SMA هایی که در دوران بچگی بروز می کند، ژن عمده ی درگیر SMN1 است.
نقشه کشی و استخراج ژن آتروفی عضلانی نخاعی
با استفاده از آنالیز پیوستگی، در سال ۱۹۹۰ هر سه شکل دوران کودکی این بیماری بر روی بازوی بلند کروموزوم ۵ تعیین نقشه گردید. تعیین جزئیات نقشه فیزیکی، لوکوس بیماری را به ناحیه ی ۱۰۰۰کیلو بازی محدود کرد. که متشکل از مضاعف شدگی وارونه ی قطعه ۵۰۰ کیلو بازی بود.
یکیاز ژن هایی که از ابتدا استخراج شد، ژن NAIP نام دار.د که پروتئین نورونی مهاری آپوپتوزیس را کد می کند. این ژن در تقریباً 45% افراد مبتلا به تیپ 1 و 20% مبتلایان به تیپ 2 و3 حذف می شود. با این وجود، برای هدف های ژنتیک مولکولی بالینی، این امر دیگر مرتبط به حساب نمی آید.
کاربردهای بالینی بیماری آتروفی های عضلانی نخاعی
آزمون تشخیصی و همچنین آزمایش ژنتیک پیش از تولد برای زوج هایی که آن را درخواست می کنند. با این فرض که هر دو حامل باشند، از اعتماد بالایی برخوردار است.
شناسایی حامل برمبنای تعیین تعداد رونوشت های ژن SMN1 که در اگزون 7 هر فرد وجود دارد، استوار است. حدود 4 درصد از جمعیت طبیعی دارای دو نسخه از SMN1 در یک کروموزوم منفرد هستند. افزون بر این، 2 درصد افراد دارای SMN1 دارای یک جهش جدید هستند، بدین معنی که تنها یک والد حامل است. به دلیل این دشواری ها، آزمون حامل SMA باید در محتوای رسمی و با مشاوره ی ژنتیک توسط متخصص کاملاً خبره ارایه شود.
مطالب بیشتر:





بدون دیدگاه